@Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways (2002)
2022-05-01: reference:
@Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways (2002) #
Their actions are opposite: while synaptic NMDAR activity induces that of CREB, eNMDAR the pathway and therefore BDNF etc.
-
Synaptic NMDA receptor activation was found to be neuroprotective.
-
Stimulation of NMDA receptors by synaptic activity robustly activated CREB and CREB target gene expression, to about the same extent as L-type calcium channels.
- Blocking L-type calcium channels with nifedipine did not reduce CRE-reporter gene expression after action potential bursts, though it did inhibit the response triggered by KClinduced membrane depolarization
- Might be relevant, idk.
- This whole deal can be replicated with Bicuculline treatment, which does not cause activation of extrasynaptic NMDAR.
- Blocking L-type calcium channels with nifedipine did not reduce CRE-reporter gene expression after action potential bursts, though it did inhibit the response triggered by KClinduced membrane depolarization
-
Glutamate treatment only poorly affected CREB and did not induce BDNF expression or TrkB phosphorylation.
- The failure of glutamate to activate CREB function was due to specific coupling of eNMDAR receptors to a CREB shut-off pathway that antagonized the CREB-promoting activity of synaptic NMDA receptors.
-
eNMDAR are composed predominantly of NR1x and NR2B. “Ifenprodil, a selective but incomplete inhibitor of NR2B may therefore preferentially inibit extrasynaptic NMDAR.”: for eNMDAR and synaptic respectively: 71% instead of 53% blocking of calcium transients for. Completely blocked CREB dephosphorylation/‘decay’. Wtf?
-
-
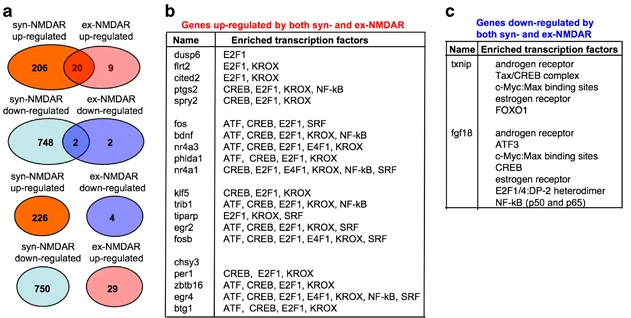
Basically, they don’t really take a stab at how their signaling cascades might differ as it relates to CREB. For now it seems to be pretty much unknown.