DNA Sequencing
2022-02-13 links: reference:
- https://en.wikipedia.org/wiki/Omics
- https://learn.gencore.bio.nyu.edu/
- https://github.com/quinlan-lab/applied-computational-genomics
- https://www.youtube.com/watch?v=yPlpVIsaRCg&list=PLCbXw1opqIQeNfF26-wWegdGoCC1aut0P&index=1
- https://www.risingtidebio.com/
Genome Sequencing #
Polymerase chain reaction (PCR) is necessary to make/amplify (millions of) copies of a DNA sample, usually done via thermal cycling In certain organisms, TACG are not the only bases in the DNA - in mammals, methyl groups may be attached, such as in 5-methylcytosine.
Analysis #
-
1% of the population needs to carry the same SNP for it to be classified as such.
- Since base pairs are always known for a strand’s corresponding one, you only need to write down one of them. (The next question is why the body even needs two?)
- Not only that, we have two copies of the DNA (we are diploids). Yes chromosomes are X-shaped pair things, but each chromosome is a pair, since we inherit one from each parent. These are refered to as homologs/homologous chromosomes. These are the ‘pairs’ we analyze and what gives rise to polymorphisms.
-
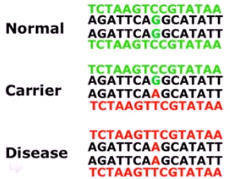 Allele simply refers to the nucleotide(s - can be umangop to hundreds) at a certain locus of interest, including of course those associated with an SNP. (Technically an SNP can have two nucleotides/alleles, and four if you count the base pairs)
Allele simply refers to the nucleotide(s - can be umangop to hundreds) at a certain locus of interest, including of course those associated with an SNP. (Technically an SNP can have two nucleotides/alleles, and four if you count the base pairs) - A genotype is the set all the alleles of a genome.
- Genotype + Environment = Phenotype
-
- Okay SNP might be able to refer to an allele that can be but may not necessarily is different.
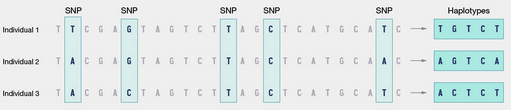
- A haplotype is basically just a set of specific nucleotides of interest that are inherited together and are usually proximal on the chromosome. Sometimes haplotypes refer to one chromosome, sometimes both.
- An SNP that leads to a different amino acid being encoded is a nonsynonymous substitution. Missense is a change of amino acid, and nonsense is a premature stop codon. Synonymous substitutions don’t change the AAs but still alter its function.
- Similarly, a silent mutation is one that does not change the codon, unlike a frameshift mutation.
- The rs number/rsID (dbSNP Reference SNP) are arbitrarily assigned I think. They correspond to specific locales and specific mutations.
For example, Acid Sphingomyelinase’s rs120074124 representing 911T>C (p.Leu304Pro (not sure what the p. stands for tbh but the AA change is the coding effect)) Niemann-Pick A.
- Sites like SNPedia/23andme have unque ‘i’ IDs.
- A haplotype is basically just a set of specific nucleotides of interest that are inherited together and are usually proximal on the chromosome. Sometimes haplotypes refer to one chromosome, sometimes both.
- At least in the context of sequencing, C/C, G/C etc. is unphased which is when you don’t know which chromosome each nucleotide is on. C|C format is phased.
- An indel is where some nucleotides are just missing.
- Tandem repeat = repeated nucleotide/set of nucleotides adjacent to eachother. Sometimes these are transcriptionally silent. There are also tandem repeat proteins, e.g. leucine-rich repeat domains, armadillo repeat domain.
- In humans, >2/3 of chromosomal DNA consists of repetitive elements; ‘repeats’. Sometimes repeats are inverted, palindromic, whatever.
- Filetypes:
- FASTQ is what’s derived from the sequencer. It’s everything.
- Alignment of a FASTQ file with a reference genome generates BAM (binary alignment map) and an associated BAI (Binary alignment index) file. From there, a VCF (variant cell format) can be produced, which is just a list of the SNPs that differ from the implied reference genome.
- There are a few standards for reference genomes, namely GRCh37 and GRCh38, which was released 4 years after the former.
- BAM can also be compressed into a CRAM file.
- I think BAM is half the size of FASTQ so I wonder if it’s just one strand or something, while both are measured on FASTQ or something.
VCF #
-
The syntax of each line is as follows:
- (#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT PRO0000283)
- chr1 11796321 rs1801133 G A 1347.77 . AC=2;AF=1.00;AN=2;BaseQRankSum=1.673;ClippingRankSum=0.000;DB;DP=45;ExcessHet=3.0103;FS=0.000;MLEAC=2;MLEAF=1.00;MQ=60.00;MQRankSum=0.000;QD=30.63;ReadPosRankSum=-0.686;SOR=0.242 GT:AD:DP:GQ:PL 1/1:1,43:44:99:1376,121,0
-
At the end is an array representing GT (genotype), AD (Allelic depths for the ref and alt alleles in the order listed), DP (Approximate read depth (reads with MQ=255 or with bad mates are filtered), GQ (genotype quality), and PL (Normalized, Phred-scaled likelihoods for genotypes as defined in the VCF specification)
- What’s obviously mostly important is GT and notice it is 1/1, indicating +/+ for the the ‘atlernate allele’, while 0 or - indicates the reference allele.
-
AC: Allele count in genotypes, for each ALT allele, in the same order as listed
-
AF: Allele Frequency, for each ALT allele, in the same order as listed
-
AN: Total number of alleles in called genotypes
-
BaseQRankSum: Z-score from Wilcoxon rank sum test of Alt Vs. Ref base qualities
-
ClippingRankSum: Z-score From Wilcoxon rank sum test of Alt vs. Ref number of hard clipped bases
-
DB: dbSNP Membership
-
DP: Approximate read depth; some reads may have been filtered
-
DS: Were any of the samples downsampled?
-
ExcessHet: Phred-scaled p-value for exact test of excess heterozygosity
-
FS: Phred-scaled p-value using Fisher’s exact test to detect strand bias
-
HaplotypeScore: Consistency of the site with at most two segregating haplotypes
-
InbreedingCoeff: Inbreeding coefficient as estimated from the genotype likelihoods per-sample when compared against the Hardy-Weinberg expectation
-
MLEAC: Maximum likelihood expectation (MLE) for the allele counts (not necessarily the same as the AC), for each ALT allele, in the same order as listed
-
MLEAF: Maximum likelihood expectation (MLE) for the allele frequency (not necessarily the same as the AF), for each ALT allele, in the same order as listed
-
MQ: RMS Mapping Quality
-
MQRankSum: Z-score From Wilcoxon rank sum test of Alt vs. Ref read mapping qualities
-
QD: Variant Confidence/Quality by Depth
-
ReadPosRankSum: Z-score from Wilcoxon rank sum test of Alt vs. Ref read position bias
-
SOR: Symmetric Odds Ratio of 2x2 contingency table to detect strand bias
Tools #
- DNA-Seq ( YT) & OakVar
- Install DNA kit studio on my windows machine?
- https://www.reddit.com/r/Nebulagenomics/comments/nhjfpa/how_to_analyze_your_own_raw_genetic_data_for_rare/
- https://usegalaxy.org/
- https://omim.org/ - catalog of genes and associated genetic disorders.
- https://www.ncbi.nlm.nih.gov/snp/?term=
- https://ancestrydna.openhumans.org/overview no idea what this is tbh besides data sharing.
- MyHeritage owns Promethease. Will have to request data deletion if I plan on using it. Apparently promethease also got fucked over by the FDA to censor results relevant to metabolism of certain pharmeceuticals or something.
-
https://www.reddit.com/r/promethease/comments/b9ls6f/super_pissed_off_promethease_removed_certain_snps/ a reddit report says they censored a SNP about fucking D2 (rs1800497) not even like some specific drug shit. That’s so retarded.
- As far as I’m aware, SNPedia is… probably not pozzed.
- But all it does its cross reference with studies on SNPedia. Maybe I should figure out how to make a FOSS alternative. That would be so amazing.
- https://codegen.eu/ has it covered I think
-
https://www.reddit.com/r/promethease/comments/b9ls6f/super_pissed_off_promethease_removed_certain_snps/ a reddit report says they censored a SNP about fucking D2 (rs1800497) not even like some specific drug shit. That’s so retarded.
- Nebula apparently uses hs38d1 reference genome as of october or november I believe.
Sequencers #
The first machine used by the Human Genome Project used the Sanger sequencing method. After that came next generation sequencers (NGS): 454, SOLiD, Illumina, MGI, DNBSEQ. Now we’re onto third-generation I think, with SMRT, Nanopore, etc.
-
Each pair is 2 bits, thus it’s 750MB total. In reality, variation is limited to <1%, so total variation can be losslessly compressed to ~4MB! Human genomes as email attachments
-
Some DNA sequences are noncoding DNA, rather serving modulatory roles.
-
https://www.labx.com/dna-sequencers It’s $8,000+ after the Ion Torrents. Nothing unique though.
- Sage Sciences?
-
The Bento Lab (nanopore) is a PCR, microcentrifuge, gel electrophoresis, and transilluminator. $1600-$2000.
Thermofisher #
They acquired Life Technologies (which also worked under the Applied Biosystems brand, which ALSO acquired SOLiD in 2006) in 2014.
-
Ion Torrent is their premier fourth-gen sequencer. https://www.thermofisher.com/us/en/home/life-science/sequencing/next-generation-sequencing/ion-torrent-next-generation-sequencing-technology.html It uses a proprietary semiconductor technology.
-
There’s like a dozen Ion Torrent PGM 508s on labx for $1000. $1500 for the 7467 (Ubuntu 10.04). idek which is newer.
- I think these fuckas are indeed from the 2000s. Search online for their names and it’s site upon site of people selling used models.
- The PGMs all take chips of either 314 (20Mb; 400-550k reads) 316 (100Mb 2-3m) or 318 (1Gb 4-5.5m) for sequence output. Read length is 200-400. >99% accurate. Single- and paired-end sequencing.
- The workflow might require o But there are plenty if you search onliner benefits from the OneTouch
-
Their modern entry-level device is the GeneStudio S5 which takes chips 510 (2-3M) up to 550 (100-130M; 25GB)
- They still don’t market this for whole genome sequencing.
- No idea how much it costs - probably $50k - it ain’t used.
- According to this flyer, the 540 and 550 chips are capable of whole genome sequencing, while for ’low-pass genome sequencing (PGS)’ the 510 is capable for 520/530 is recommended.
Illumina #
GC content bias describes the dependence between fragment count (read coverage) and GC content found in Illumina sequencing data
HiSeq, MiSeq, HiScanSQ, Genome Analyzer IIx, etc. They have entry-level machines but according to them, the only ones capable of human whole-genome sequencing is the big as NovaSeq 6000.
Oxford Nanopore #
- You need to make a nanopore account. https://store.nanoporetech.com/us/sample-prep.html I dont’t know what 99% of this does.
MinION #
-
Despite the ION name, look at Nanopore - because that’s the technology they use, which utilizes flowcells: often some combination of microfluidics, high precision optics and electronics; sequencing reagents are at a minimum highly pure natural biochemicals like A/G/C/T-TP, but more typically such molecules modified in all sorts of interesting ways and used with highly engineered versions of natural enzymes or other proteins.
-
The flowcell measures electrical changes caused by DNA being driven through protein pores in membranes.
-
As of 2016, the minION Mk1b provided 2.4x coverage and 8.7Gb with one flow cell over 48 hours. Afaik, it’s dead for good after 48 hours. So, maybe you get 2 good uses out of it?
- As of 2022 it can generate up to 50Gb. Flow cells are $900 and can be stored at 2-8°C for up to 12 weeks.
- $100 wash kits allow them to be reused up to 5 times:
https://store.nanoporetech.com/us/flow-cell-wash-kit-r9.html. By the hour, the ‘pore complex’ deteriorates, decreasing the read rate over time.
- Software allows one to check the number of nanopores (n=512) remaining.
- $100 wash kits allow them to be reused up to 5 times:
https://store.nanoporetech.com/us/flow-cell-wash-kit-r9.html. By the hour, the ‘pore complex’ deteriorates, decreasing the read rate over time.
- As of 2022 it can generate up to 50Gb. Flow cells are $900 and can be stored at 2-8°C for up to 12 weeks.
-
Also required is the actual library kit, which is looking like $600 minimum maybe?
-
MinION Mk1B: $1000
- 50Gb per flow cell when run for 72 hours at 420 bases/second.
- Conect to a computer. Recommended specs are a 4-core i7, and a 2060 with 8GB VRAM. R. The software used is MinKNOW, Guppy the basecaller, and EPI2ME for data analysis.
-
MinION Mk1C:
- AIO device with a touchscreen and pre-installed basecalling and analysis software.
-
New method helps pocket-sized DNA sequencer achieve near-perfect accuracy
- Using a barcoding system (I assume it’s one of those several hundred $ kits) the MinION can be incredibly accurate.
Sequence Assembly #
- The devices must assemble contigs, fragments of 20-30,000 bases depending on the reading capability of the device. IT’s kinda speculative how many genes we have, but it’s something like 30,000-50,000, with about 3 billion base pairs. Bottom-up sequencing shears DNA into small fragments and reassembling them based on overlaps, up to the entire genome. The alignment is done via software: AMOS is an open source deal but this might be a dead project - but it may be all is needed.