
It’s April 2022 in a niche nootropics community, and the next potential superdrug has been discovered. Does the power it may hold have the potential to change the entire world as we know it?

-
Heavy metal chelator, especially 3+ ions (Ferric Iron, Aluminum, lead, mercury, chromium, zinc, copper, etc.) limited affinity for ferrous iron.
- Deferoxamine has a very high affinity and specificity for the ferric iron and chelates it in a 1:1 molar ratio; that is, 100 mg of deferoxamine will bind to and eliminate 8.5 mg of elemental iron
- Binds free iron in the bloodstream as well as intralysosomal ferritin/siderin. Does not readily bind iron from transferrin, hemoglobin, myoglobin, or cytochrome (not typo).
-
 Fe-DFO = ferrioxamine B, which is water soluble and is excreted in the kidneys and feces. Half-life of ~10 hours.
Fe-DFO = ferrioxamine B, which is water soluble and is excreted in the kidneys and feces. Half-life of ~10 hours. - Al-DFO = aluminoxamine, which increases blood aluminum concentrations. According to pubchem, 100mg dfo can bind ~4.1mg aluminum.
-
AFAIk, it targets or at least effects every organelle: cyotosol, endolysosome, etc. However it isn’t membrane-permeable? I really want to know what this means for its pharmcokinetics?
-
Ferritin accumulates as a consequence of endolysosomal deacidification promoting expression of Ferritin Heavy Chain. Failure of ferritin turnover within our braincells leads to the decline of the autophagesome and defective autophagy that’s present in alzheimers, down syndrome, Autism sprectrum disorder, Parkinsons, Huntingtons disease, nathman pick disease, etc.
- This makes sense, since H+-ATPase mediates the acidification, and if respiration is deficient then there’s no ATP, immediately creating a deadly cycle.
- Iron leaks out of mitochondria dislodging from the mitochondrial machinery and into the LIP whereby it generates ROS.
- DFO should rescue ferritin turnover, and shuttle it towards mitochondria if needed. In certain disorders i.e. where autophagy is compromised, the iron in ferritin simply doesn’t leave.
- Utilization alongside EPO andor ferritin pore unfolding peptide:Apart from Lipid rafts, extracellular atp, and loss of neural stem cells and senescence this would spell the end for neurodegeneration
- We can target inflammatory purine receptors with p2x7 inhibition, reduce degeneration of our lipid rafts by promoting insulin sensitivity, reducing sphingolipid incorporation with inhibitors, and promoting their breakdown with glucocerebosidase activators. We promote better liver health which reduces ammonia, and bilrubin accumulation that aggravates endozapine release and further damages the lipid rafts of neuronal cells. Then we can increase our neuronal stem cell pools by increasing insulin sensitivity and activation with insulin/IGF2 and reducing oxidative stress with powerful mitochondrial targeted antioxidants.
-
Lysosomal deacidifcation also increases iron intake into hepcidin and ferritin.
-
As we age, the activity of Heme Oxygenase increases as a means of attempting to mitigate oxidative stress and that breaks down hemoglobin into bilrubin, carbon monoxide, and free iron.
- So the process which iron continues to accumulate within the brain becomes more aggressive and this is partially responsible for the dysfunction of Erythropoietin activity and increased levels of epo within the brain in Alzheimer’s or Parkinson’s disease. Epo can’t exert its functions on mitochondria promoting mitochondrial biogenesis and it also leads to the breakdown of neuronal insulin sensitivity and ferritin turnover.
-
In CSF, 5-HT level is significantly decreased and the levels of iron and transferrin are dramatically increased in fatigue group
-
iron dysbiosis even leads to the failure of phageocytosis of microglial cells (THEIR degradation, pretty sure) and that means that microglial cells can nolonger properly break down neuronal progenitor cells that dont incorporate into neuronal networks or consume dead cells and clearance of associated cellular wastes. Iron dysbiosis even supports the survival and reactivation of latent bacteria within the brain that increase inflammation through endotoxemia and it all feeds forward
-
The mitochondria in this instance is called the Endosome; what’s outside of mitochondria where all other organelles of our cells are is called the lysosome
-
Potent induction of angiogenesis can make it a topical for Hair Loss?
-
Covid is a hepcidin memetic. Severity has been shown to be related to how well it is metabolized, i.e. in younger people.
-
What I want to find out is its cellular localization and its dynamics on mitochondria and cellular respiration. Main concerns really if you look at good/ugly.
-
- Cellular iron depletion elevated protein levels of the early endosomal marker Rab5.
The Good
-
Significantly inhibits BACE1
-
Deferoxamine: a reversible S-phase inhibitor of human lymphocyte proliferation
- Potent inhibitor of DNA synthesis by T Cells and B Cells Prevents cells from completing the S phase of the cell proliferation cycle.
-
- 50 mg/kg/d, and initial elimination half-life of 0.28/h and steady-state concentration of 7μM/L were observed.
-
Increased iron levels in the lung reduced function and worsened pulmonary fibrosis, and was prevented with DFO. Lung fibrosis enters the iron age
-
- Deferoxamine ‘induces autophagy’, which leads to Ferritin entry into Lysosomal lumen.
- DFO-treated cells resulted in cytosolic accumulation of LC3B, while DFX/DFP, or ferrioxaxmine, did not.
- Incubation of DFO-treated cells with 3-methyladenine, an autophagy inhibitor, resulted in degradation of ferritin by the proteasome.
- This process of feritin turnover is important for rechanneling iron into the proteome which autophagy-promiting compounds favor.
- The membrane-permeable chelators desferasirox (DFX) and deferiprone diverted ferritin degradation towards the proteasomal pathway.
- Did not induce pH changes in the lysosome.
- Inhibition of endocytosis prevents DFO-induced ferritin degadation, supporting the importance of the lysosomal localization of Deferoxamine.
- Furthermore, it might be specific to siderophores or sometihng, because another impermeable chelator, bathophenanthroline did not induce autophagy.
- Rapamyci] can induce macroautophagy but does not induce Ferritinophagy.
- Some kinda PI3K inhibitors inhibit lysosomal ferritin degradation.
- our results show that ferritin can be degraded either by the lysosome or the proteasome
- Lysosomal deferoxamine can induce Ferritinophagy even when entry of ferritin into the lysosome is prevented.
- A combination of DFO and a permeable chelator provides a shuttle mechanism by which DFO takes iron from the permeable chelator for it to again bind more iron.
- Deferoxamine ‘induces autophagy’, which leads to Ferritin entry into Lysosomal lumen.
-
A recent study has shown that DFO and DFX increased autophagy via inhibiting mTOR. Their differences may be cell-type dependent.
-
The hypoxia memesis is by means of… for one, removing Fe2+ from Prolyl Hydroxylase, which marks HIF-1α and HIF-2α for ubiquination.
- Stimulation of HIF-1alpha, HIF-2alpha, and VEGF by prolyl 4-hydroxylase inhibition in human lung endothelial and epithelial cells
- Prolyl Hydroxylase inhibition enhances VEGF release and glucose consumption
- Stimulation of HIF-1alpha, HIF-2alpha, and VEGF by prolyl 4-hydroxylase inhibition in human lung endothelial and epithelial cells
Insulin
- iron accumulates in astrocytes, reduces insulin sensitivity and that prevents insulin increasing the transcription and prevents the release of Erythropoietin within the brain.
- This would support why neuronal levels of epo decline and systemic levels of epo rise with age or could even support why neuronal epo might be higher in dementia as a consequence of failure of epo to exert its actions on mitochondrial biogenesis due to the rise of hepcidin, and ferritin within the cytosol of neurons, and microglial cells
- Tl;dr, DFO fixes this by lowering GSK, thereby Tau. Increases HIF and thereby insulin signaling.
- Iron Deposition Leads to Hyperphosphorylation of Tau and Disruption of Insulin Signaling
- Via decreased phosphorylation levels of IR-β (unable to detect changes in levels) IRS-1, and PI3K.
- Treatment of insulin within a short time led to a rapid and transient hyperphosphorylation of tau
- Cognition is impaired and causes abnormal tau phosphorylation in mice fed high-iron chow.
- Iron Depletion by Deferoxamine Up-Regulates Glucose Uptake and Insulin Signaling in Hepatoma Cells and in Rat Liver
Neuronal
-
Reversion of age-related recognition memory impairment by iron chelation in rats
- Neonatal iron supplementation induces selective accumulation in certain brain regions, notably the basal ganglia, associated with memory impairments in adulthood. It also induces lipid peroxidation and protein carbonylation in substantia nigra.
-
- Neuroinflammation from Endotoxins etc. activates Microglia release of TNF-α, IL-1β, etc. This leads to:
- Hepcidin expression is increased in astrocytes/microglia
- Ferroportin is decreased in all 3 cell types.
- DMT1 expression is increasd in all 3 cell types
- So, iron accumulates in neurons and microglia (but not astrocytes) with hepcidin binding to iron otherwise present in mitochondria.
- Aging is associated with increased brain iron through cortex-derived hepcidin expression
- Here, we measured the levels of iron in different tissues of aged mice, and demonstrated that while cytosolic non-heme iron is increased in the liver and muscle tissue, only the aged brain cortex exhibits an increase in both the cytosolic and mitochondrial non-heme iron
- Aging is associated with increased brain iron through cortex-derived hepcidin expression
- Neuroinflammation from Endotoxins etc. activates Microglia release of TNF-α, IL-1β, etc. This leads to:
-
- Iron chelators pick up redox-active iron… would that mean only Fe2+?
- 100μM pentupled Lactate Dehydrogenase acivity.
- Not too much erythropoiesis. 500μM doubled.
-
- Deferoxamine-induced TH upregulation (and DAT upregulation apparently) was attenuated by ERRγ inverse agonist.
-
Neuroprotective effect of deferoxamine on erastin-induced ferroptosis in primary cortical neurons
- Inhibits the ferroptotic pathway by upregulating the Cystine-Glutamate Antiporter system light chain (xCT) and Glutathione Peroxidase 4.
-
- Upregulated Tyrosine Hydroxylase. Downregulated DMT1+IRE and TfR1
- Inhibited MPTP-induced phosphorylation of JNK; enhanced phosphorylation of ERK and p38.
-
- Chelation via DFO decreased the stability of DAT mRNA, increasing its phosphorylation and ubiquination. Increased its localization into the endosome.
- Rab5 increased 3-fold and Rab11 decreased 5-fold.
- Protein Kinase A -62%, Protein Kinase B -81%, Protein Kinase Cγ -32%. Protein Kinase Cδ -60%, and Protein Kinase Cζ -40%, but Protein Kinase D increased 250%.
- Down-regulation of dopamine transporter by iron chelation in vitro is mediated by altered trafficking, not synthesis
-
- Both in vitro and in vivo, FHC upregulation was accompanied by loss of mature dendritic spines, which was also dependent on μ-Opioid Receptor and Gαi-protein signaling.
- Deferoxamine blocked morphine-mediated FHC upregulation. Iron chelation with Deferoxamine (100 µM, 24 h) did not significantly change basal FHC or FLC expression
- The same morphine treatment significantly increased cytoplasmic iron levels as measured by phen green SK, which was blocked by chelating endolysosomal iron with DFO
- Morphine’s ability to reduce dendritic spine density was blocked by chelation of endolysosomal Iron with Deferoxamine, but not affected by extracellular iron chelation with DTPA, demonstrating the importance of endolysosomal iron for this pathway. Morphine-mediated reduction of mature dendritic spines requires endolysosomal iron. This is massive. At the end of the day, DFO blocked morphine-induced neurological dysfunction.
- morphine and FAC significantly reduced thin and mushroom spines, and this effect was similarly blocked by DFO, but not DTPA;
- Morphine and FAC both significantly reduced overall dendritic spine density by the same amount. Morphine’s ability to reduce dendritic spine density was blocked by chelation of endolysosomal iron with DFO.
- Both in vitro and in vivo, FHC upregulation was accompanied by loss of mature dendritic spines, which was also dependent on μ-Opioid Receptor and Gαi-protein signaling.
-
Lysosomal iron modulates NMDA receptor-mediated excitation via small GTPase, Dexras1
- NMDAR activation can induce Iron movement into neurons via DMT1 (Divalent metal transporter), and under pathological conditions like excitotoxicity, contributes to metal-catalyzed ROS generation.
- S-nitrosylation of Dextras1 by Nitric Oxide induces DMT1.
- Genetic and pharmacological ablation of this neuronal iron pathway (decreasing cytosolic iron) in the mice increased glutamatergic transmission and synaptic plasticity, due to synaptic modification of NMDA receptor activity via modulation of the PKC/Src/NR2A pathway.
- Lysosomal iron serves as a main source for intracellular iron signaling modulating glutamatergic excitability
- In wild type CA1 slices (no toxicity no nothing) 80% increase in spontaneous EPSC after application of pyridoxl isonicotinoyl hydrazine, a membrane-permeable iron chelator.
- NMDAR activation can induce Iron movement into neurons via DMT1 (Divalent metal transporter), and under pathological conditions like excitotoxicity, contributes to metal-catalyzed ROS generation.
-
Translocation of iron from lysosomes into mitochondria is a key event during oxidative stress‐induced hepatocellular injury pretty interesting stuff actually. Just read it for localization info.
- ROS formation initiates mitochondrial permeability transition (MPT) that opens nonspecific permeability transition pores in the Inner Mitochondrial Membrane These pores conduct all solutes up to 1500 Da. Sounds like suicide!
- Es gibt ein non-chelatable iron pool in ferritin, proteins, Fe-S clusters, etc. that chelators like deferoxamine cannot reach - only free iron, and iron bound less strongly to anions.
- DFO is poorly permeable, not impermeable.
- Evidence that desferrioxamine cannot enter cells by passive diffusion
- Deferoxamine enters cells by endocytosis, and then localizes to lysosomes/endosomes.
- What transporter takes it up??
- Evidence that desferrioxamine cannot enter cells by passive diffusion
- Mitochondria accumulate Fe2+ electrogenically via the mitochondrial Ca2+ uniporter, while Fe3+ is not transported.
-
Reactions of desferrioxamine with peroxynitrite-derived carbonate and nitrogen dioxide radicals
- Prevents peroxynitrite-mediated oxidations and attenuates Nitric Oxide and oxygen radical-dependent oxidative damage.
Amyloid
-
- Induced M2 activation of Microglia and inhibited M1 activation, in the hippocampus of APP mice.
- This means TGF-β→AKT
- 0.56-fold Caspase 3.
- Induced M2 activation of Microglia and inhibited M1 activation, in the hippocampus of APP mice.
-
Aβ is capable of accumulating Iron (III) within amyloid aggregates, with this process resulting in Aβ-mediated reduction of iron(III) to a redox-active iron(II). The presence of Aluminum increases the reductive capcity.
The Bad
-
- This paper is about mitochondrial trafficking. That’s important for arborization and outgrowth as they go from old to new areas I suppose
- At 11 days in vitro, DFO reduced average mitochondrial speed (kinesin/dynein and adaptor proteins) by increasing the pause frequency of individual dendritic mitochondria. Time spent in retrograde motion was reduced.
- We have previously shown that chronic iron deficiency (ID) impairs mitochondrial respiration and dendritic complexity without severely impairing overall neuronal health: Iron Deficiency Impairs Developing Hippocampal Neuron Gene Expression, Energy Metabolism, and Dendrite Complexity
-
Deferoxamine deconditioning increases neuronal vulnerability to hemoglobin This is after blood vessel rupture, which causes an elevation in tissue iron. 3-4x neuronal loss after Hemoglobin exposure, since it releases iron upon Heme Oxygenase.
-
- Similar compounds with nitroxyl groups/hydroxylamines are known to produce nitric oxide under oxidative conditions. DFO incubation with sheep adult/fetal blood resulted in formation of HNO into Nitric Oxide, forming iron nitrosyl hemoglobin. This is still just under oxidative conditions, with ferricyanide. NO is only generated when DFO is injected directly with FeCN.
- The reaction does not happen in pure deionized water. Spontaneously nitrite formation is observed in DFO + $\ce{CO3^{2-}}$ even without FeCN.
- Reducing agent $\ce{I3^-}$ didn’t lead to any NO generation.
- DFO at >1 mM concentrations can produce a nitroxide radical via oxidation by a hydroxyl radical produced by photolysis of H2O2.
- NO generation was eliminated upon DFO pre-treatment with anaerobically prepared FeSO4
- Production of NO from DFO+FeCN was not affected by pre-treatment with 100 μM H2O2, contrary to expectations.
- Plasma concentrations of DFO are reported to reach between 80 and 130 μM following 3 min of intravenous injection
- Similar compounds with nitroxyl groups/hydroxylamines are known to produce nitric oxide under oxidative conditions. DFO incubation with sheep adult/fetal blood resulted in formation of HNO into Nitric Oxide, forming iron nitrosyl hemoglobin. This is still just under oxidative conditions, with ferricyanide. NO is only generated when DFO is injected directly with FeCN.
-
- Its stability constant of 10^31 for ferric iron contrasts significantly with those for other ions such as zinc (10^11), calcium (10^2) and magnesium (10^4).
- Since certain compounds liberate iron from ferritin, iron excess can occur without ‘iron overload’.
- Ferritin, lipid peroxidation and redox-cycling xenobiotics
- Oxygen-based free radical generation by ferrous ions and deferoxamine
- DFO accelerates autoxoidation of Fe2+ into Fe3+, associated with uptake of oxygen; fenton reactions, and with a fall in pH.
- DFO may directly scavenge $\ce{OH^.}$ and $\ce{O2^{.-}}$.
- DFO conjugated with starch (purpose is simply to increase molecular weight, I think) is unable to penetrate into RBC (as if DFO wasn’t impermeable enough), and decreases NAD redox potential. Thus this effect probably is borne of the extracellular realm, yet the NAD is inside the erythrocyte.
- 500μL of whole blood with 0-6mM. That’s crazy compared to the ~10μM/L in clinical use in vivo. Not bad for a 15% drop in redox potential.
- Able to induce oxidation of ferrous iron? They say it’s likely to be involved but I fail to recognize how it wouldn’t just be spontaneous
-
- Its permeability is favored by its positive charge.
- Its uptake into hepatocytes is 800x faster into red blood cells at 37°C.
- Tommy: Causes a rebound in non-Tf-bound iron if delivered at high doses.
- Removes ~1/3 of non-Tf-bound iron rapidly, and increased dosing does not increase this removal, instead leading to greater rebound on cessation.
- Technique used is stabilizing DFO with aluminum ions so as to prevent non-Tf-bound iron shuttling to it after collecting plasma.
The Ugly
- Deferoxamine Enhanced Mitochondrial Iron Accumulation and Promoted Cell Migration in Triple-Negative MDA-MB-231 Breast Cancer Cells Via a ROS-Dependent Mechanism
- Under iron-deficient conditions, increased mitochondrial iron levels in these cancer cells increases ROS generation. DFO is an anticancer drug - I think this ROS is a good thing or some shit.
- Cell penetrating peptide (CPP)-conjugated desferrioxamine for enhanced neuroprotection: synthesis and in vitro evaluation
- The applications of DFO are limited because of its inability to access intracellular labile iron. Honestly, WTF? Where does it go then?
- This is a lie. I mean, maybe it doesn’t touch it, but it certainly decreases LIP: R
- Its limited permeability is due to high hydrophobicity and high molecular weight.
- Bidentate (3:1 stabilizing iron) ligands are likely to be toxic.
- The applications of DFO are limited because of its inability to access intracellular labile iron. Honestly, WTF? Where does it go then?
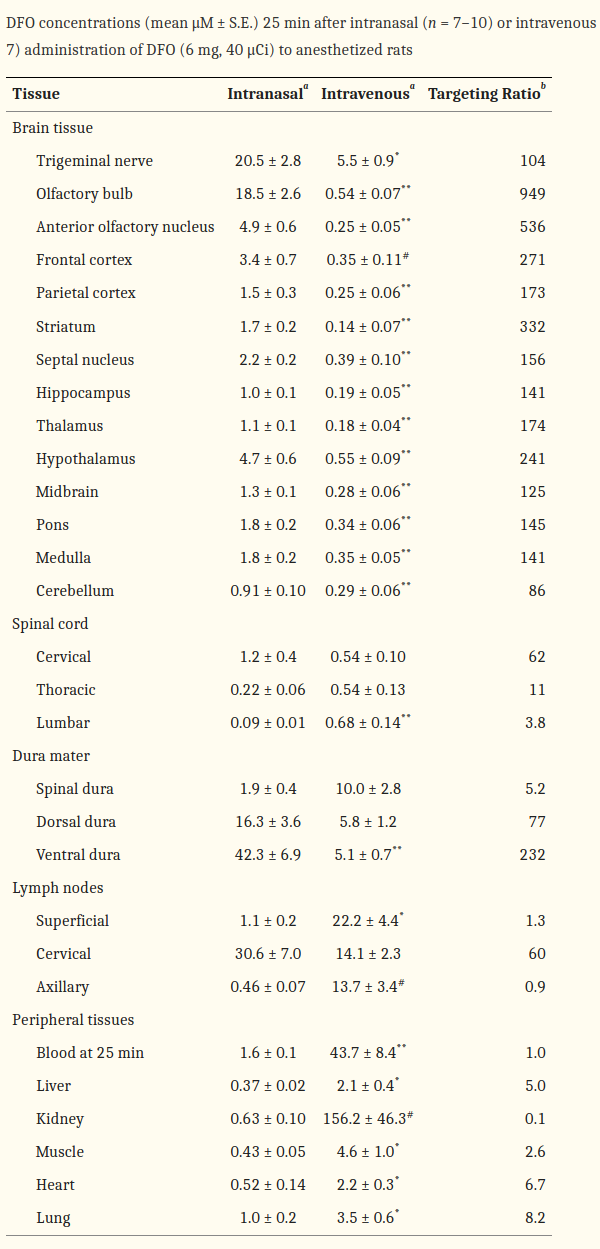
Intranasal studies
-
Mechanisms of Intranasal Deferoxamine in Neurodegenerative and Neurovascular Disease
-
Intranasal Administration of Deferoxamine to Iron Overloaded Patients
-
[Intranasal deferoxamine improves performance in radial arm water maze, stabilizes HIF-1α, and phosphorylates GSK3β in P301L tau transgenic mice (Fine 2012)]
-
- I mean this is still an Alzheimer’s model that reverses the consequences of NFTs. Nonethleess it increased morris water maze performance before streptozotocin treatment.
-
- Both pGSK-3β and β-Catenin were significantly increased by approximately 50% in the DFO-treated mice
-
- Not only are they healthy, but these are 7 week old mice. That’s like 5 year old kids. The iron accumulation has hardly begun! Though, perhaps, maybe neither has natural chelation from perhaps high prenatal levels (if that even happens).
- DFO may slow the process of retinal degeneration (Obolensky et al., 2011) and improve aspects of human cerebrovascular function, namely vasoreactivity and autoregulation, from baseline (Sorond et al., 2015).
- 100mg/kg = ~600 mg HED. It was administered daily for over a month! 18g would be nuts mate…
- Significantly decreases GSK-3β activity via phosphorylation at Ser9. In this study: 68% increase in HIF-1α, 200% increase in pGSK3β, ~40% increase in β-Catenin. Contrary to expectations, ~30% decrease in GLUT1.
- Treatment results in phosphorylation of AKT. Along with VEGF I can think of, yet another mechanism to decrease GSK-3β. The inhibition of it is neuroprotective in brain injury.
- Decreased Amyloid β and Tau in AD, and preservation of striatal DA neurons in PD.
- Chronic intranasal deferoxamine ameliorates motor defects and pathology in the α-synuclein rAAV Parkinson’s model
- Iron can compromise the solubility of α-synuclein. DFO did not protect against Parkinson’s model dopaminergic cell death, but it decreased the number of pathological α-synuclein formations at the terminal level.
- Lack of weight loss or ‘physical abnormalities’.
- Intranasal Deferoxamine Provides Increased Brain Exposure and Significant Protection in Rat Ischemic Stroke
-
- Serial Studies of Auditory Neurotoxicity in Patients Receiving Deferoxamine Therapy
- An analysis of Jhe clinical data showed that most members Of the affected group were younger, had lower serum ferritin value, and were receiving higher doses of the drug per kilogram of body weight. These people are 3-27 years, mean 12.
- *Most of these reports had dealt with a variety of ocular changes ranging from blurring of vision [5], loss of acuity (4,6-81, loss of central vision [4], night blindness [9], pigmentary retinopathy [4,9,10], and optic neuropathy [4,9,11]. *
- Most cases of acute visual loss were reversible after stopping therapy with deferoxamine, but anatomic abnormalities such as optic neuropathy and pigmentary changes remained unchanged.
- In our four symptomatic patients [4], two presented with a marked decrease in central vision, eccentric fixation, and severely impaired visual acuity. The symptoms completely reversed within four weeks after withdrawal of deferoxamine, but optic atrophy and thinning of the nerve fiber layer persisted. A third patient had markedly decreased visual acuity, a left afferent pupillary defect, and asymmetric optic atrophy: after withdrawal of the drug, acuity improved but the atrophy was unchanged. The fourth patient also had decreased visual acuity, abnormal color vision, and translucent swelling of the optic disk; after stopping the drug, acuity and color vision partly improved. Abnormal pigmentary changes were observed in five other patients [4], but there were no abnormalities of visual acuity or color vision in these cases.
Other chelator considerations
Deferiprone
- A Combined Drug Treatment That Reduces Mitochondrial Iron and Reactive Oxygen Levels Recovers Insulin Secretion in NAF-1-Deficient Pancreatic Cells
- Memebrane-permeable baby. And coadministrated with NAC.
Dosage
Lasts reconstituted for 2 days.