The following is a literature review that got out of hand after reading Cholesterol in Alzheimer’s disease: unresolved questions. Hopefully you’ll find some of the points interesting! This definitely has everything you need to get started, at least.
-
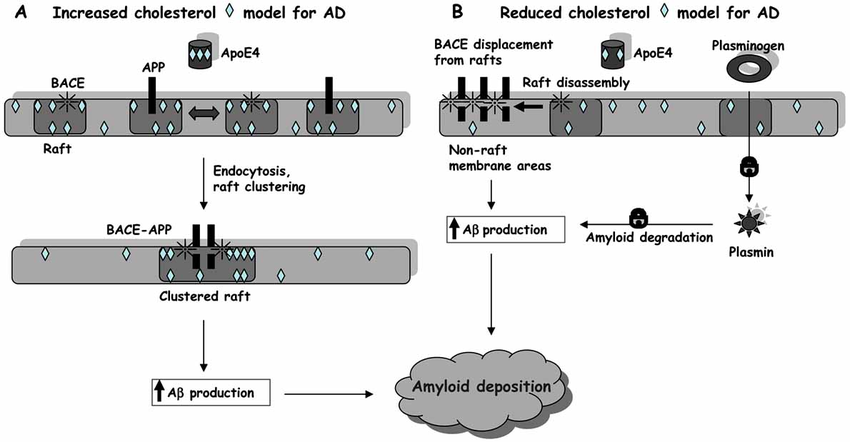
So apparently there are two opposing models: The ‘high cholesterol model’ assumes the presence of cholesterol→lipid rafts allows for colocalization of APP and BACE1.
- cf. the entire ‘lipid raft signaling hypothesis’: Lipid raft microdomains and neurotransmitter signalling, i.e the idea that (caveolar) lipid raft microdomains form kinetically favorable interactions for protein complexes, or rather, perhaps they inhibits them by separating. Or both, of course.
-
The low cholesterol model assumes APP is located in non-raft membrane regions, with lipid rafts keeping it separated from BACE1. Furthermore, they note that activation of plasminogen to plasmin is a raft-associated event, and a reduction would somehow increase the poroduction of amyloid for some reason.
-
So, seems to me that this is essentialy predicated on where APP likes to associate, which could just be due to its chemical structure or whatever, which is yet another thing that makes me wonder about its differences with AC-rER or other peptides.
-
LRP1 is essential for ApoE endocytosis+catabolism. LRP1 expression seems to be under negative transcriptional control of APP or its intracellular domain: Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1
-
Most of the brain cholesterol is translocated to the circulation across the BBB in the form of the oxidized derivative 24S-hydroxycholesterol. This is a measure of Cholesterol turnover in the CNS, since cholesterol 24-hydroxylase is responsible for ~40% of cholesterol turnover/catabolism in the brain. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. 27OH-Chol originates from the periphery and crosses the BBB - so, vascular damage will see decreased CNS 24S-hydroxycholesterol and increased 27OH-Chol, which is seen in late AD (in early AD, 24SOH-Chol increases in CSF and periphery, reflecting increased brain cholesterol turnover.)
High Cholesterol Model
- Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts
- BACE1 seems to require intact rafts for activity, and BACE1 outside of rafts appears to be inactive.
- They demonstrated a decrease of cellular cholesterol inhibited Aβ generation, consistent with something like Cholesterol-dependent formation of GM1 ganglioside-bound amyloid beta-protein, an endogenous seed for Alzheimer amyloid : Increases in not only GM1 but also cholesterol contents in the lipid bilayers facilitated the binding of Abeta to the membranes by altering the binding capacity but not the binding affinity.
- GM1/Aβ is never found in the normal brain, only in those exhibiting early pathological changes of AD
- Generation of GM1/A b is facilitated by the combination of cholesterol and sphingomyelin
- Enrichment of cholesterol of the host membranes facilitated the generation of GM1/Aβ via formation of a GM1 “cluster” that acts as a binding site of Aβ.
- γ-Secretase is also lipid raft associated: Gamma-secretase activity is present in rafts but is not cholesterol-dependent
- An explanation for Wahlre 2002’s report that it is cholesterol-dependent is not clear. Perhaps: they treated the cells with a γ-secretase inhibitor prior to harvest. Using the parent APP-transfected CHO cells, two of the present authors (H.M. and Y.I.) initially made an observation suggesting that the cellular cholesterol levels can modulate γ-cleavage of APP. When the cells were treated with 25-hydroxycholesterol, which reduces the levels of cholesterol and elevates those of Sphingomyelin, the levels of intracellular Aβ‚ significantly decreased, while those of β-CTF increased.
- This ultimate effect is very similar to that of a potent γ-secretase inhibitor. However, we found no suppression of Aβ production by membranes isolated from 25-hydroxycholesterol-treated cells. Thus, these data raise the possibility that the treatment of in vitro cultured cells with a certain inhibitor prior to cellular fractionation may perturb additional cellular especially lipid) parameters to compensate for aberrant metabolism, and as a result thus-prepared membranes may not necessarily represent the membrane under normal metabolic conditions. In other words, the γ-secretase activity would be modulated or influenced by many factors that may be involved mostly in membrane integrity.
- An explanation for Wahlre 2002’s report that it is cholesterol-dependent is not clear. Perhaps: they treated the cells with a γ-secretase inhibitor prior to harvest. Using the parent APP-transfected CHO cells, two of the present authors (H.M. and Y.I.) initially made an observation suggesting that the cellular cholesterol levels can modulate γ-cleavage of APP. When the cells were treated with 25-hydroxycholesterol, which reduces the levels of cholesterol and elevates those of Sphingomyelin, the levels of intracellular Aβ‚ significantly decreased, while those of β-CTF increased.
- However, most of these data were obtained in cells heavily perturbed by chemical treatment with methyl-β-cyclodextrin or lovastatin. The former extracts cholesterol from the plasma membrane, thus heavily affecting lipid rafts, and is a general endocytosis inhibitor, whereas statins have been shown to alter APP processing by modifying cell cholesterol distribution, rather than total levels, with its increase in the exofacial membrane leaflet.
- Cholesterol distribution, not total levels, correlate with altered amyloid precursor protein processing in statin-treated mice (Burns et al. 2006)
- All three statins caused CNS cholesterol to translocate from the cytofacial leaflet to the exofacial leaflet! (AKA intra vs. extracellular-facing)
- There might be differences in expression between the two: [Cholesterol—A Janus-Faced Molecule in the Central Nervous System] says exofacial has PC, SM, PE, and cytofacial has PE, PS, and PI.
- Only lovastatin significantly reduced total cholesterol in isolated plasma membranes.
- Nontransgenic mice. Reduced Aβ40/42 and decreased β-CTF.
- All three statins caused CNS cholesterol to translocate from the cytofacial leaflet to the exofacial leaflet! (AKA intra vs. extracellular-facing)
- Referencing: Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10 (Kojiro et al.)
- “See commentary: A fluid connection: Cholesterol and Aβ”
- ADAM10 associates with membranes that are cholesterol-poor, like those with Phospholipids.
- These guys were pretty influential as well: Simvastatin strongly reduces levels of Alzheimer’s disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo (Fassbender et al. 2001). This has caveats being some kind of megaddose though
- Did not reduce cholesterol, but did reduce lathosterol (the precursor to cholesterol) and Aβ by about 50%. The reduction of Aβ occurring in absence of any change in cholesterol could be explained by a minor cholesterol compartment in neurons that changes more rapidly, but whose size is too small to be reflected in measures of total brain cholesterol. Alternatively, it is also possible that the critical species regulating Aβ production is another lipid in the cholesterol biosynthetic pathway.
- Kojiro observed no significant increase in α-secretase until the reduction in cholesterol production is greater than 50%.
- Although α-secretase activity is a separate activity than Aβ production, this result raises the possibility that Aβ will decrease only when a threshold of cholesterol reduction is achieved. If this possibility is true, then why did simvastatin reduce Aβ production despite little, if any, change in total cholesterol levels? The answer might be that a precursor of cholesterol regulates Aβ production in vivo.
- The former, right?
- Although α-secretase activity is a separate activity than Aβ production, this result raises the possibility that Aβ will decrease only when a threshold of cholesterol reduction is achieved. If this possibility is true, then why did simvastatin reduce Aβ production despite little, if any, change in total cholesterol levels? The answer might be that a precursor of cholesterol regulates Aβ production in vivo.
- Sites of APPsα production occur in membrane regions with low cholesterol content and high fluidity
- Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease
- After cholesterol depletion, a 3-6x increase in APPsα was observed.
- “See commentary: A fluid connection: Cholesterol and Aβ”
- Nonetheless this study still recognizes that APP happens on the cell surface, and that cholesterol-rich caveolae microdomains may play a role in a-secretase mediated proteolysis of PAPP in vivo: Caveolae, plasma membrane microdomains for alpha-secretase-mediated processing of the amyloid precursor protein
- Alpha secretase processing was significantly promoted by recombinant overexpression of caveolin.
- ApoE promotes cholesterol and phosphatidylcholine! efflux from astrocytes/neurons.
- Lovastatin increases ADAM10 expression by unknown mechanisms!
- Cholesterol distribution, not total levels, correlate with altered amyloid precursor protein processing in statin-treated mice (Burns et al. 2006)
- [Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons (Simons 1998)]
- Cholesterol decreases secretion of the secreted form of amyloid precursor protein by interfering with glycosylation in the protein secretory pathway
- Ganglioside GM1 efficiently decreases the membrane microviscosity
- Re-assessing the relationship between cholesterol, statins and Alzheimer’s disease (Wolozin et al. 2006)
- Statins inhibit isoprenylation as it is an extension of the cholesterol biosynthetic pathway (from Farnesylpyrophosphoric acid), and it is required for APP processing (via ras, rac, rho.).
- Clinical studies suggest that high doses of statins can reduce Ab load, but the same clinical studies suggest that statins delay the progression of AD rather than preventing the incidence of AD. Our own studies using neuropathology raise the possibility that statins prevent the progression of AD by inhibiting inflammation.
- Elevated dietary cholesterol uptake increased amyloid plaque formation in rabbits and transgenic mice:
- Similarly: Alzheimer’s disease: the cholesterol connection
- A hallmark of Alzheimer’s is an abnormal accumulation of Aβ. Nay, its accumulation is the initiating factor.
- Elevated cholesterol levels increase Aβ in cellular and most animals models of AD, and drugs that inhibit cholesterol synthesis lower Aβ in these models.
- The identification of a variant of the Apolipoprotein E gene as a major genetic risk factor for AD is also consistent with a role for cholesterol in the pathogenesis of AD.
- The Mevalonate Pathway in Alzheimer’s Disease — Cholesterol and Non-Sterol Isoprenoids BIG thing on Cholesterol synthesis!
- down-regulation of the mevalonate pathway may play an important role in the increased rates of cognitive decline in AD. Studies at the subcellular level suggest that SREBP-2 may be posttranslationally regulated in AD. We demonstrated that oAβ42 inhibit SREBP-2 maturation in cultured neurons. We also discovered that the levels of (M)SREBP-2 are reduced in the frontal cortex of the AD CRND8 mouse
Low Cholesterol Model
-
The beneficial effects of low brain cholesterol resulting from this model are strongly questioned by several studies carried out on experimental systems close to the physiological and pathological situations. Actually, many data suggest that cholesterol loss in neuronal membranes enhances amyloid peptide generation.
- The conflicting role of brain cholesterol in Alzheimer’s disease: lessons from the brain plasminogen system (2005)
- It has not been shown that higher neuronal cholesterol increases Abeta production… it has not been demonstrated that neurons in AD have more cholesterol than control neurons. On the contrary, the brains of AD patients show a specific down-regulation of Seladin-1, a protein involved in cholesterol synthesis, and low membrane cholesterol was observed in hippocampal membranes of ApoE4 (apolipoprotein E4) AD cases.
- What would you know, there is controversy on seladin-1’s role.
- In female mice, the most brain-permeant statin induces neurodegeneration and high amyloid production
- It appears more likely that the advantageous role of statins arises from improved brain oxygenation
- It has not been shown that higher neuronal cholesterol increases Abeta production… it has not been demonstrated that neurons in AD have more cholesterol than control neurons. On the contrary, the brains of AD patients show a specific down-regulation of Seladin-1, a protein involved in cholesterol synthesis, and low membrane cholesterol was observed in hippocampal membranes of ApoE4 (apolipoprotein E4) AD cases.
- Plasma membrane cholesterol controls the cytotoxicity of Alzheimer’s disease AβP (1–40) and (1–42) peptides
- PC12 cells become resistant to the cytotoxic action of AβP when incubated in a medium that enriches cholesterol levels of the surface membrane. On the other hand, making cholesterol-deficient membranes by either cholesterol extraction with cyclodextrin or by inhibiting de novo synthesis of cholesterol makes PC12 cells more vulnerable to the action of Aβ
- Increasing cholesterol content of PS liposomes also suppresses Aβ-dependent liposome aggregation
- The conflicting role of brain cholesterol in Alzheimer’s disease: lessons from the brain plasminogen system (2005)
-
Neuronal membrane cholesterol loss enhances amyloid peptide generation (2004)
- Much higher levels of BACE 1–APP colocalization is found in hippocampal membranes from AD patients or in rodent hippocampal neurons with a moderate reduction of membrane cholesterol!
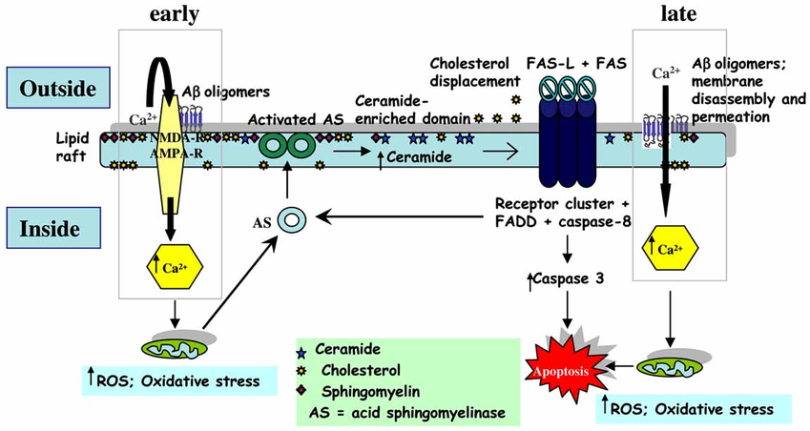
- The ultimate question is where cholesterol plays a role in this causality. It’s no mystery that increased ceramides play a role. But what about ganglioside?
- γ-Secretase executes an intramembranous cleavage, which is apparently difficult and uncommon. They compare it to SREBP and Notch which are also intramembraneous, and indeed the linkage is kind of directly especially with the latter.
- See:
- See:
- Much higher levels of BACE 1–APP colocalization is found in hippocampal membranes from AD patients or in rodent hippocampal neurons with a moderate reduction of membrane cholesterol!
-
- According to previous data and our, reduced cholesterol level in plasma membrane and intracellular compartments by the deficiency of DHCR24 activity obviously was involved in β-amyloid generation, tau hyperphosphorylation, apoptosis.
- Universally regulated by sterols/steroids, ACTH, thyroid, neurotrophins, and xenobiotics.
- Apparently it is no paradox that impaired cholesterol catabolism and trafficking leads to a loss in brain cholesterol!
Etc. Notes
- “I believe that the reduced secretion of sappa from the nucleus due to a failure in autophagesome formation may be more reflective of the phenotype observed in sappa overexpression models or in autism”
- Here we have Insulin→mTORC2 and overall ceramide accumulation which can be thanks to abberant Acid Sphingomyelinase from an Aβ feedback loop, ROS, or a feedback loop in general from ceramide displacing cholesterol.
- Alzheimer’s Disease as a Membrane Disorder: Spatial Cross-Talk Among Beta-Amyloid Peptides, Nicotinic Acetylcholine Receptors and Lipid Rafts
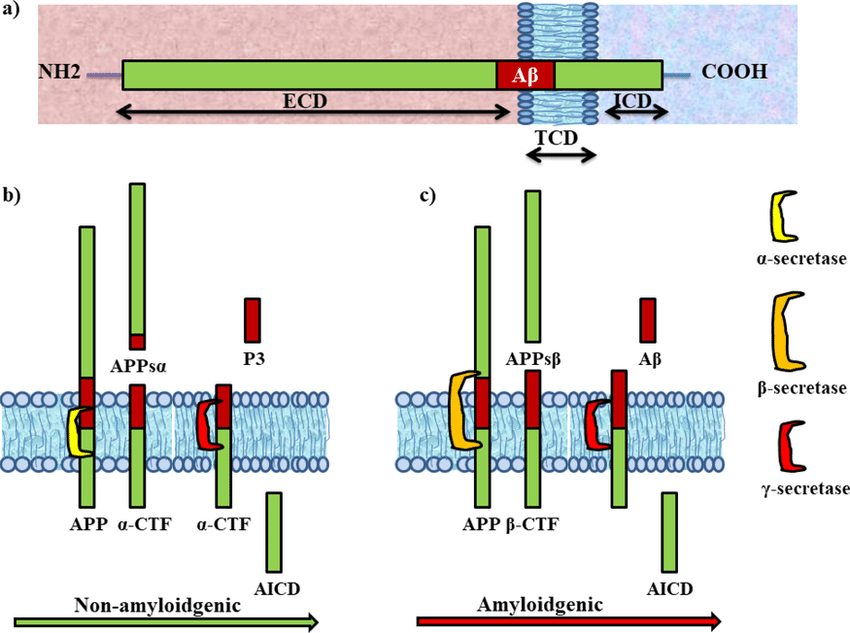
- a) β-Secreatase is present in both raft and non-raft domains, but needs to be in raft domains to be functional.
- b) β-secretase in raft domains corresponds to an incative pool that needs to relocate to non-raft domains to be functional.